
Die krankhafte Neubildung von Blutgefäßen im Auge ist ein zentraler Faktor bei Erkrankungen der Netzhaut wie der diabetischen Retinopathie oder der altersbedingten Makuladegeneration, die zu einem dauerhaftem Sehverlust und Erblindung führen können. Ein internationales Forscherteam unter der Leitung von Professor Dr. Jens Kroll von der Medizinischen Fakultät Mannheim der Universität Heidelberg hat nun einen bislang unbekannten molekularen Mechanismus aufgeklärt, der zur krankhaften Neubildung von Blutgefäßen im Auge beiträgt.
Die Wissenschaftler vermuten, dass dies ein Ansatzpunkt für neue, zielgerichtete Therapien sein könnte. Da es bereits für andere Erkrankungen zugelassene Medikamente gibt, die an diesem Mechanismus angreifen, erscheint dieser Ansatz als sehr vielversprechend. Die Ergebnisse der internationalen Studie wurden aktuell im Fachjournal Nature Communications veröffentlicht.*
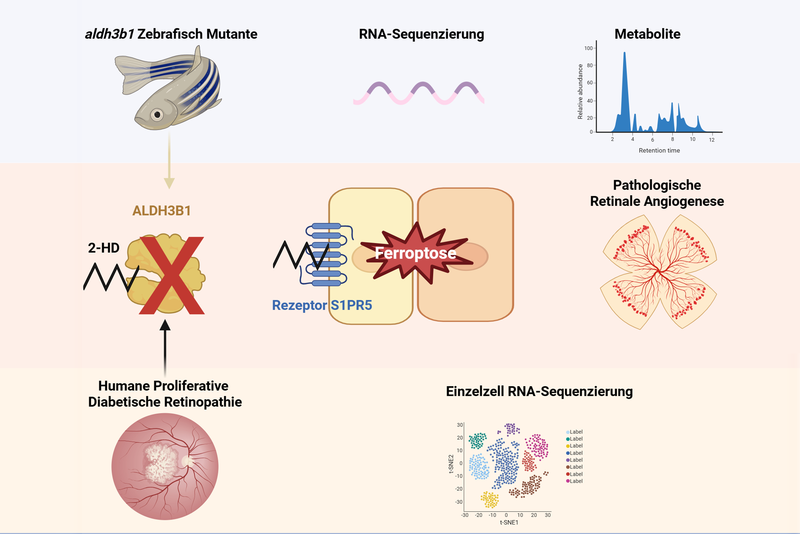
Im Mittelpunkt steht Sphingosin-1-Phosphat (S1P), ein Botenstoff aus der Gruppe der Sphingolipide, der eine Schlüsselrolle in der Regulation von Gefäßwachstum, Entzündungsprozessen und Gewebereparatur einnimmt. Während die Funktionen von S1P selbst bereits gut untersucht sind, war bislang unklar, ob auch seine Stoffwechselprodukte eine Rolle spielen könnten. Die Forscher konnten nun zeigen, dass ein kleines reaktives Lipidaldehyd namens 2-Hexadecenal (2-HD) eine entscheidende Rolle bei der Entstehung krankhafter Gefäßneubildung spielt.
Neue Einblicke in die Pathogenese von Augenerkrankungen
Für ihre Forschung nutzten sie das Zebrafischmodell und Daten von Patienten mit krankhaften Neubildungen von Blutgefäßen im Auge. Um die Funktion von 2-HD zu untersuchen, schalteten die Forscher ein Enzym (ALDH3b1) aus, das 2-HD „entgiftet“. Fische, denen ALDH3b1 fehlte, entwickelten spezifisch im Auge ein Gefäßmuster, das dem späten Stadium der Netzhauterkrankungen im Menschen ähnelte. Dieser Prozess war umkehrbar, wenn die Wirkungen von 2-HD geblockt wurden. Als Schalter identifizierten sie den S1P-Rezeptor 5 (S1PR5), einen von fünf bekannten S1P-Rezeptoren, die sich auf der Oberfläche von Immunzellen finden. S1PR5 wird gezielt von 2-HD gehemmt, was zur Fehlregulation der Gefäßbildung führt.
Durch die Kombination von Genaktivitätsprofilen und detaillierten chemischen Analysen konnten die Forscher auch auf den zugrundeliegenden Mechanismus schließen. Sie fanden heraus, dass ein Überschuss an 2-HD den Eisenhaushalt der Zellen störte und sie in Richtung Ferroptose trieb, einer eisenbedingten Form des Zelltods, der durch eine außer Kontrolle geratene Lipidperoxidation gekennzeichnet ist. Diese pathologischen Prozesse tragen direkt zur Schädigung der Blutgefäße bei, die die Netzhaut des Auges versorgen.
Die Ergebnisse liefern erstmals einen direkten Zusammenhang zwischen einem S1P-Metaboliten und der Regulation von Gefäßwachstum im Auge. Damit erweitern sie das bisherige Verständnis der molekularen Grundlagen von Gefäßerkrankungen des Auges erheblich.
Potenzial für neue Therapieansätze
Aktuelle Therapien, etwa die Hemmung des vaskulären endothelialen Wachstumsfaktors (VEGF), sind zwar wirksam, weisen jedoch Einschränkungen wie Nebenwirkungen oder die Notwendigkeit wiederholter invasiver Behandlungen auf. Die neu identifizierte Signalachse rund um 2-Hexadecenal und S1PR5 eröffnet alternative medikamentöse Therapieansätze, die entweder über die gezielte Hemmung von 2-HD wirken oder über die Stabilisierung des S1PR5-Rezeptors. Dabei ist besonders vielversprechend, dass für die Behandlung der Multiplen Sklerose S1PR5-Rezeptor Modulatoren im klinischen Alltag eingesetzt werden, es also bereits zugelassene Medikamente gibt, die an diesem Mechanismus ansetzen.
*Publikation
Qian, X., Ge, R., Chu, Y. et al.
Sphingosine-1-Phosphate-derived 2-Hexadecenal is a central mediator of ocular neovascularization by inhibiting Sphingosine-1-Phosphate receptor 5.
Nat Commun 17, 3488 (2026).
DOI: https://doi.org/10.1038/s41467-026-71792-3
Kontakt
Prof. Dr. med. Jens Kroll
Medizinische Fakultät Mannheim der Universität Heidelberg
ECAS/Abteilung Vaskuläre Biologie
E-Mail: jens.kroll@medma.uni-heidelberg.de